质子交换膜水电解(PEMWE)是生产绿色氢气的重要方法,但其阳极析氧反应(OER)动力学缓慢,电催化剂在强酸、高电位下易腐蚀降解。目前,铱(Ir)基催化剂虽稳定性好、活性较高,但储量少、成本昂贵,限制了PEMWE的发展。二氧化钌(RuO2)因成本较低且本征活性高,被视为理想的替代品,然而在高电位下 Ru 位点易过度氧化形成可挥发性或可溶性物种,导致催化剂溶解失活。 为提高 RuO2在酸性环境中的稳定性,可将反应机制从晶格氧机制(LOM)转向吸附物演化机制(AEM)。AEM 涉及四个连续的质子耦合电子转移步骤,在传统 RuO2上这些步骤集中于单一Ru位点,去质子化困难,尤其在富质子的酸性电解液中更受限。引入额外质子受体(如通过异质金属取代)可活化桥氧原子,加速氧中间体的去质子化,但该方法会减少 Ru 活性位点,并可能引入阳离子蚀刻,加剧膜降解。因此,如何在保持 Ru 活性位点总数不变的前提下,有效调控桥氧辅助的去质子化过程,仍是当前面临的重大挑战。
中国科学技术大学吴长征教授、中国科学院深圳先进技术研究院彭晶副研究员等研究人员开发了一种氟阴离子调控策略,赋予RuO2高效稳定的酸性水氧化性能。F-诱导的适度氢键相互作用加速了氧中间体(以及水)的去质子化过程,从而改善了析氧反应(OER)动力学。此外,高电负性的F-削弱了Ru-O键的共价性,从而提高了RuO2催化剂的长期稳定性。值得注意的是,RuO1.86F0.14催化剂在10 mA cm-2电流密度下表现出极高的活性,过电位仅为153 mV ,优于大多数已报道的酸性条件下的电催化剂。此外,该催化剂在10 mA cm-2电流密度下稳定运行超过980小时,且降解速率仅为27 μV/h。当应用于PEMWE时,RuO1.86F0.14催化剂在1.63 V的电池电压下可提供1 A cm-2的电流密度,并在60 °C下保持超过100小时的稳定性。原位光谱表征技术和理论计算表明,氢键介导机制(HBM)在氟化RuO2的析氧反应(OER)过程中占主导地位。其中,吸附中间体(或水)上的质子通过氢键自发转移到相邻的桥氧上,导致快速去质子化过程,并抑制了晶格氧介导的反应路径。
相关研究成果以“Accelerated Deprotonation Triggered by Fluorinated RuO2 Enables Efficient and Stable Acidic Water Electrolysis”为题发表在J. Am. Chem. Soc.上。

氟离子触发的氢键介导去质子化新机制:通过引入氟离子,在RuO2表面形成氢键网络,加速了氧析出反应(OER)中间体的去质子化过程。该机制有效降低了酸性环境中质子转移的能垒,显著提升了OER动力学。
活性与稳定性的双重提升:氟的高电负性削弱了Ru–O键的共价性,抑制了Ru位点的过氧化和溶解。在保持Ru活性位点数目的前提下,实现了催化剂在酸性介质中的高活性与长时稳定性(>980 h)。
阐明稳定性机理:结合多种表征技术,证实氟化RuO2主要遵循吸附演化机制(AEM),而非晶格氧机制(LOM),从而抑制了结构降解。DFT计算显示氟诱导的氢键网络降低了反应决速步能垒,加速了质子转移。
实际PEMWE器件中展示优异性能:RuO1.86F0.14在质子交换膜水电解槽中仅需1.63 V即可实现1 A cm-2的电流密度,并稳定运行超过100小时。该催化剂表现出与商业Ir基催化剂相当的效能,具备工业化应用潜力。
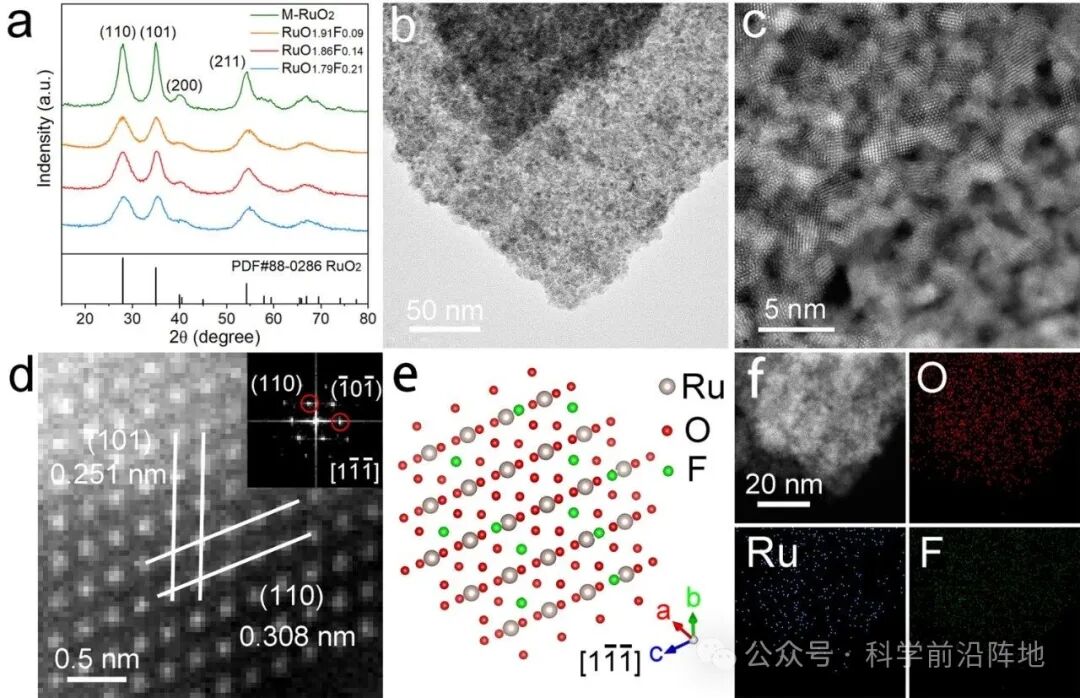
图1. RuO1.86F0.14催化剂的合成路线及结构表征。(a) 合成样品的XRD图谱。(b) RuO1.86F0.14催化剂的TEM图像。(c) RuO1.86F0.14催化剂的HAADF-STEM图像。(d) RuO1.86F0.14催化剂的HAADF-STEM放大图像及FFT图谱(插图)。(e) RuO1.86F0.14沿[11̅1̅]方向的晶体结构模型。(f) RuO1.86F0.14催化剂中Ru、O和F元素的EDS元素映射图像
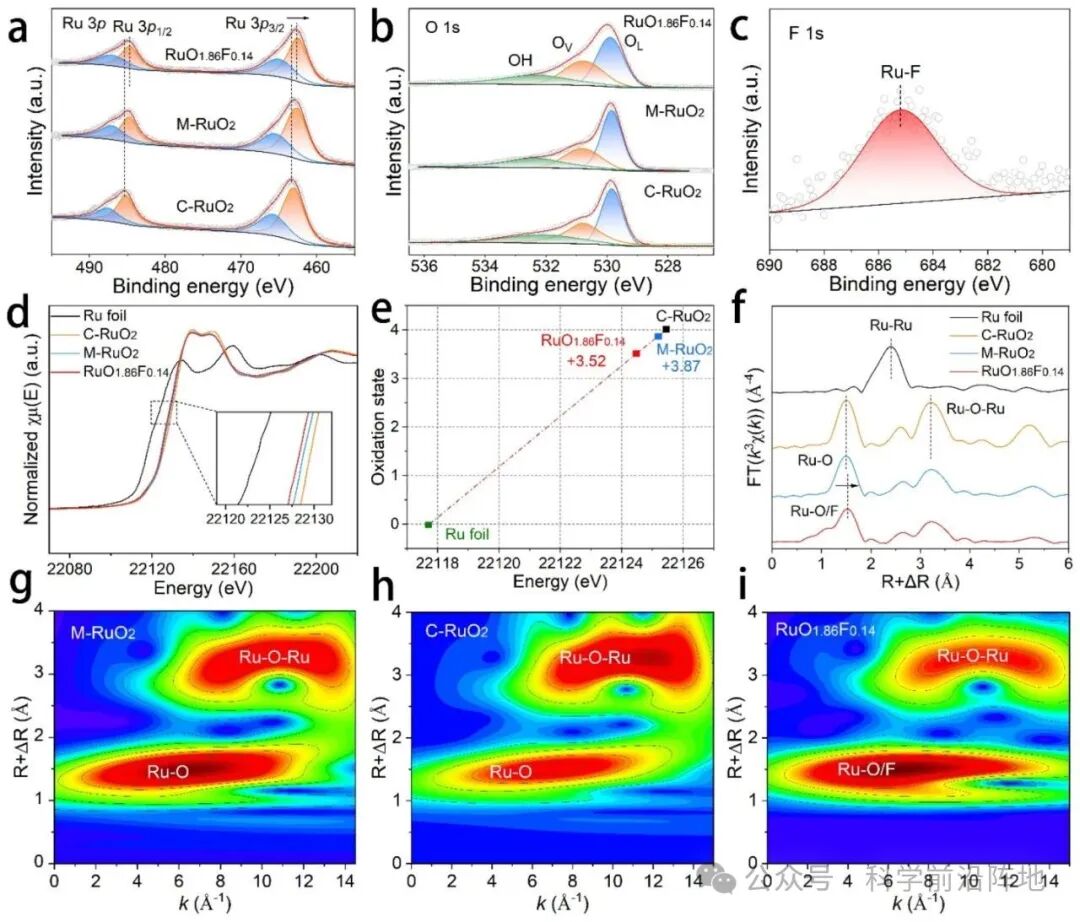
图2. RuO1.86F0.14催化剂的电子结构表征。(a) RuO1.86F0.14、M-RuO2和C-RuO2的Ru 3p XPS谱图。(b) RuO1.86F0.14、M-RuO2和C-RuO2的O 1s XPS谱图。(c) RuO1.86F0.14的F 1s XPS谱图。(d) Ru箔、C-RuO2、M-RuO2和RuO1.86F0.14的Ru K边XANES谱图。插图:XANES谱图放大图。(e) 由Ru K边XANES谱图确定的各种钌物种的氧化态。(f)Ru箔、C-RuO2、M-RuO2和RuO1.86F0.14的k3加权Ru K边EXAFS光谱的傅里叶变换。(g-i)M- RuO2、C-RuO2和RuO1.86F0.14的k3加权Ru K边EXAFS光谱的小波变换

图3. 电催化析氧反应(OER)性能测试。(a) 不同催化剂在5 mV s-1扫描速率下,90% iR补偿的线性扫描伏安法(LSV)曲线。 (b)不同催化剂在10 mA cm-2电流密度下的过电位和在1.53 V vs RHE下的质量活性比较。 (c) RuO1.91F0.09、RuO1.86F0.14、RuO1.79F0.21、M-RuO2和C-RuO2的Tafel斜率。 (d) RuO1.86F0.14、M-RuO2和C-RuO2在10 mA cm-2电流密度下的计时电位曲线。(e)在酸性介质中,10 mA cm-2电流密度下,RuO1.86F0.14与最近报道的Ru基OER催化剂的降解速率和稳定性比较
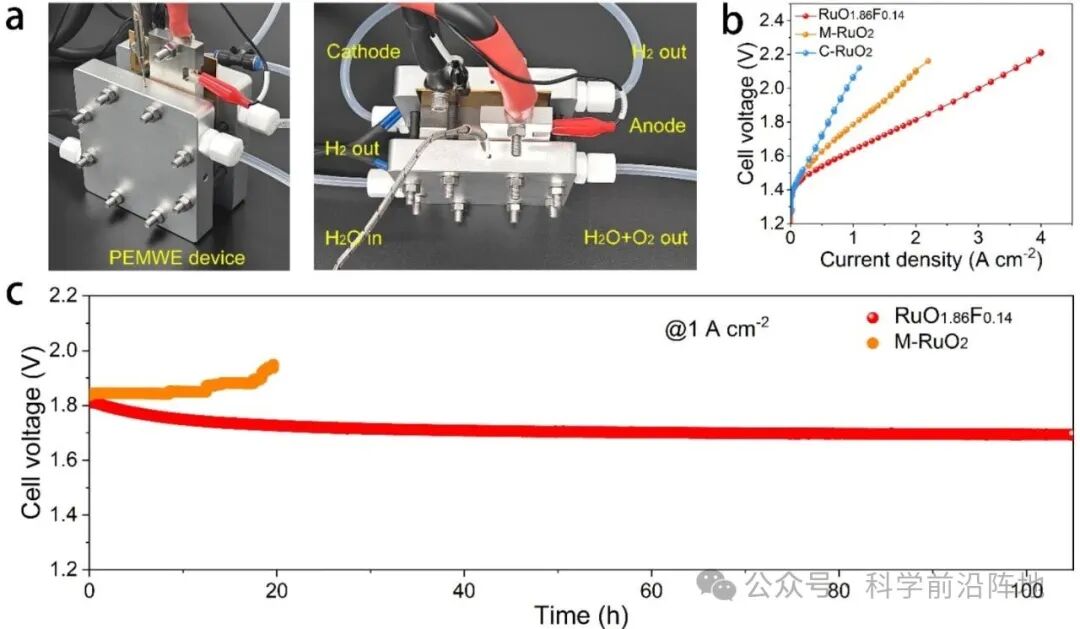
图4. (a) 电解槽装置的照片。(b) 以RuO1.86F0.14和C-RuO2为阳极催化剂的PEMWE在60 °C下的极化曲线。(c) 以RuO1.86F0.14为阳极催化剂的PEMWE在1 A cm-2和60 °C下的计时电位曲线
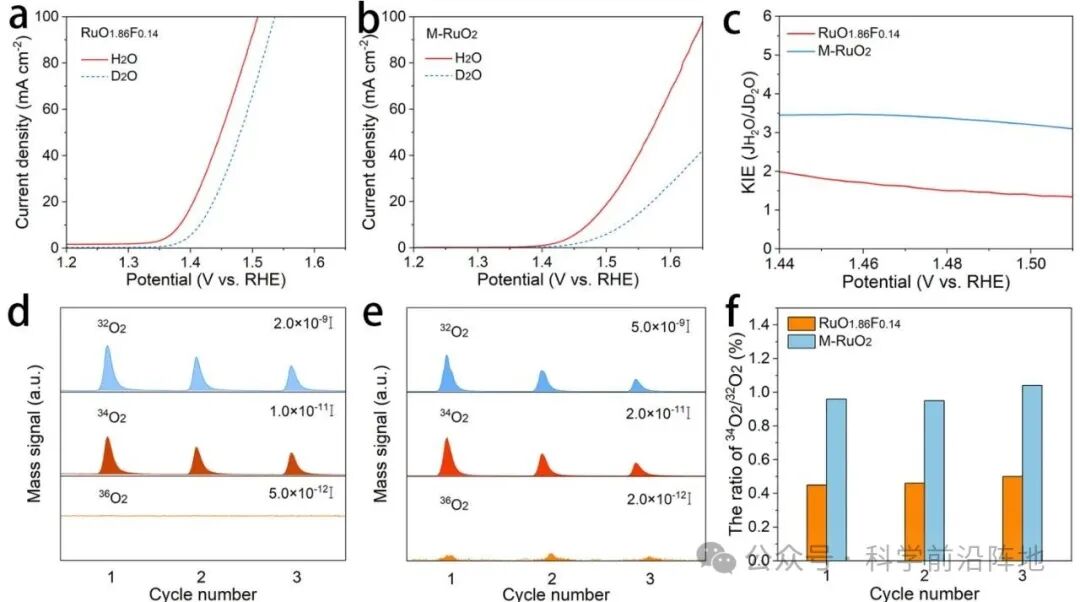
图5. RuO1.86F0.14的析氧反应(OER)机理。(a) RuO1.86F0.14和(b) M-RuO2在含H2O或D2O的0.5 M H2SO4溶液中的线性扫描伏安(LSV)曲线。(c) 不同电位下获得的动力学同位素效应(KIE)值。 (d)和(e)分别为18O标记的RuO1.86F0.14和M-RuO2催化剂在含H2O的0.5 M H2SO4电解液中三个循环过程中气态产物中32O2、34O2和36O2的DEMS信号。 (f)各循环伏安(CV)循环中M-RuO2和RuO1.86F0.14的34O2/32O2比值
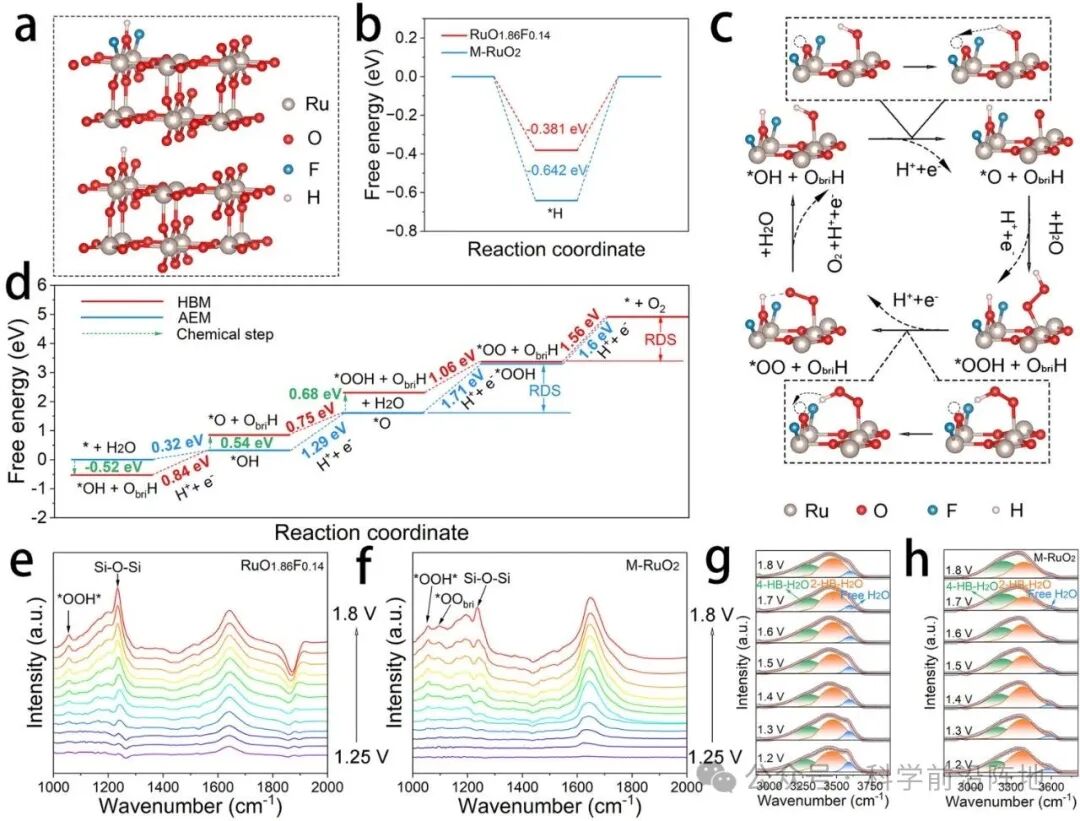
图6. 电子结构的 DFT 计算和 OER 过程的原位光谱研究。(a) RuO1.86F0.14和M-RuO2(110)表面的吸附氢结构。(b) 不同催化剂表面Obri位点的氢原子吸附能。(c) RuO1.86F0.14(110)表面上通过所提出的HBM路径的关键中间体演化过程示意图。(d) RuO1.86F0.14表面上不同OER路径的自由能图。(e) 和 (f) 酸性OER过程中不同施加电位下RuO1.86F0.14和M-RuO2的原位衰减全反射表面增强红外吸收光谱(ATR-SEIRAS)。利用原位ATR-SEIRAS技术,研究了酸性析氧反应(OER)过程中不同外加电位下RuO1.86F0.14(g)和M-RuO2(h)界面水的结构。强氢键水、弱氢键水和非氢键水分子分别通过拟合的O-H伸缩振动峰表示。图(g)和(h)中的绿色、橙色和蓝色峰分别对应于4-氢键水、2-氢键水和游离水
这项研究通过引入氟阴离子对RuO2进行电子结构调控,不仅提出并证实了一种全新的“氢键介导去质子化”机制,显著提升了酸性氧析出反应的动力学,更重要的是,该策略在不牺牲活性位点的前提下,通过削弱Ru–O键共价性从根本上抑制了活性中心过氧化与溶解,从而实现了高活性与高稳定性的统一。这一工作将催化剂的理性设计从传统的阳离子掺杂扩展到阴离子工程,为破解酸性水电解中贵金属催化剂“活性-稳定性”权衡这一长期困境提供了创新思路,同时通过原位光谱与理论计算的深度融合,为揭示复杂电催化界面反应的真实路径树立了范例,对开发下一代高性能、低成本的质子交换膜电解水制氢技术具有重要的科学指导与工程应用价值。
原文链接:
https://pubs.acs.org/doi/10.1021/jacs.5c08859
声明:仅代表作者个人观点,作者水平有限,如有不科学之处,请在下方留言指正!

来源|科学前沿阵地
